Gaussian¶
简介¶
Gaussian是一个用于计算化学、分子和量子化学计算的软件套件。它主要用于从头开始计算分子的电子结构和能量,以及模拟分子的行为。Gaussian提供了各种量子化学计算方法,包括从简单的哈特里-福克 (Hartree-Fock)方法到更高级的密度泛函理论(Density Functional Theory, DFT)方法。
Gaussian在科研和工业界都得到了广泛的应用,用于研究分子结构、反应机理、光谱性质等。它通常由化学家、物理学家和生物学家等专业人士使用。
安装环境¶
序号 |
集群 |
平台 |
版本 |
位置 |
安装方式 |
|---|---|---|---|---|---|
1 |
hpckapok1 |
Cpu |
g16 |
/share/install_packages/g16.tar.gz |
个人安装 |
2 |
hpckapok2 |
Cpu |
g16 |
/public/source/g16.tar.gz |
个人安装 |
使用方法¶
软件安装¶
注意
Gaussian软件使用权限属于组或个人,需要把软件解压至个人目录再运行.
hpckapok1¶
cp -r /share/install_packages/g16.tar.gz ~
# 解压软件到个人目录
tar -zxvf g16.tar.gz
cd g16
./bsd/install
#创建临时目录
mkdir ~/tmp
hpckapok2¶
#安装软件到个人目录
cp /public/source/g16.tar.gz ~
tar -zxvf g16.tar.gz
# 创建临时目录
mkdir ~/tmp
slurm作业提交¶
注意
需要先把软件解压至个人目录再运行.
提前创建临时目录.
1.编写Gaussian.slurm脚本
#!/bin/bash
#SBATCH -J STDIN_0118_141938
#SBATCH -p cpuXeon6458
#SBATCH -N 1
#SBATCH --ntasks-per-node=1
#SBATCH --time 03:00:00
#SBATCH --comment=BASE
#SBATCH --array=1-1 ### MARK_MULTI_SUB
#!/bin/bash
# 设置软件环境变量
export g16root=~
export GAUSS_EXEDIR=$g16root/g16
export GAUSS_SCRDIR=~/tmp
export PATH=$PATH:$g16root/g16
source $g16root/g16/bsd/g16.profile
# 开始计算
g16 <测试文件路径>
2.使用 sbatch Gaussian.slurm 提交作业
web平台提交¶
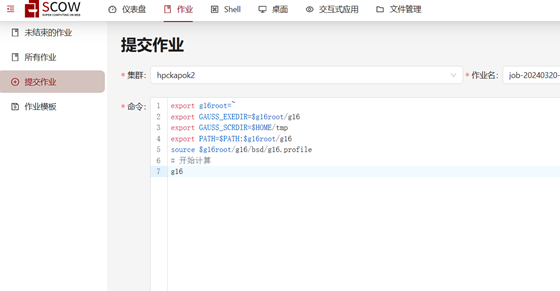
1.在“提交作业”页面填写作业信息
命令行使用¶
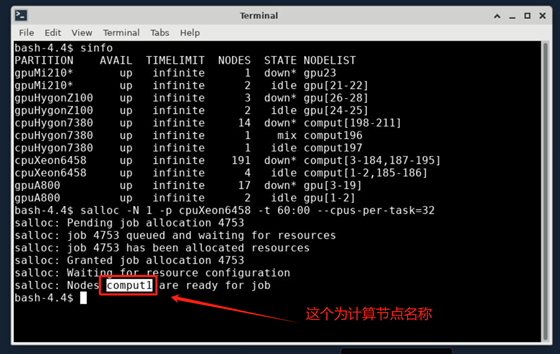
1.在终端申请计算节点资源,申请资源命令如下:
# 申请资源命令例子,申请一个节点、cpuXeon6458资源分区、使用时长60分钟、32核
salloc -N 1 -p cpuXeon6458 -t 60:00 --cpus-per-task=32
命令行参数解释:
-N <节点数量>
--cpus-per-task=<单进程 CPU 核心数>
--gres=gpu:<单节点 GPU 卡数>
-t <最长运行时间>
-p <使用的分区>
--qos=<使用的 QoS>
2.根据分配的计算节点,使用ssh -Y <计算节点>登录至计算节点。例如下图,分配的节点为comput1,则命令为: ssh -Y comput1
3.登录至节点并启动软件
# 登录至节点
ssh -Y <计算节点名称>
# 设置软件环境变量
export g16root=~
export GAUSS_EXEDIR=$g16root/g16
export GAUSS_SCRDIR=~/tmp
export PATH=$PATH:$g16root/g16
source $g16root/g16/bsd/g16.profile
# 开始计算
g16 <测试文件路径>
软件测试方法¶
1.根据 slurm作业提交 章节,发起任务提交作业
2.计算结算后,使用 formchk 任务结果.chk转化成fchk格式文件
参考资料¶
论文致谢模板¶
(中文)本研究工作得到得到了华南理工大学科学计算公共服务平台的支持;
(英文)This work is partially supported by High Performance Computing Platform of South China University of Technology.
Contributor:B君