节点资源选择¶
在集群上执行自己的并行作业之前,需要找到每个作业的最佳节点数(-n)、任务数(--ntasks-per-node,--nstasks/-n)、CPU核心数(--cpus-per-task/-c)等参数,在某些情况下还需要选择合适的GPU数量。 本页演示和分析不同参数情况下的作业表现,以帮助我们找到设置这些参数的最佳值。
cpu和gpu使用率¶
首先要考虑运行脚本能利用好申请的资源。
提交脚本后,用squeue获取申请到的计算节点。
$squeue
然后ssh登录到计算节点,最右侧标记为“NODELIST(REASON)”的列提供运行作业的计算节点的名称。
$ssh NODENAME
登录成功后,用top -u $username(你的用户名) 命令查看cpu使用率。对于gpu卡,使用nvidia-smi或者watch -n 1 nvidia-smi动态查看gpu使用率。
多线程cpu利用率¶
申请资源如下:
#SBATCH --nodes=1
#SBATCH --cpus-per-task=16
cpu利用率如图所示
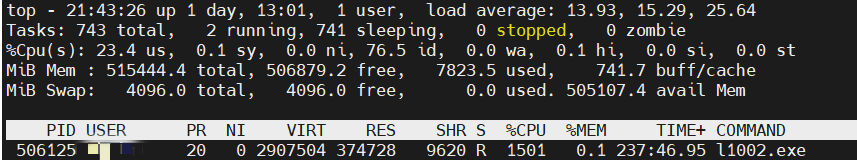
图中,PID 506125 CPU使用率为1501%,说明使用了多线程,16个核跑满了。
并行作业的cpu利用率¶
申请资源如下:
#SBATCH --nodes=1
#SBATCH ---ntasks-per-node=16
cpu利用率如图所示
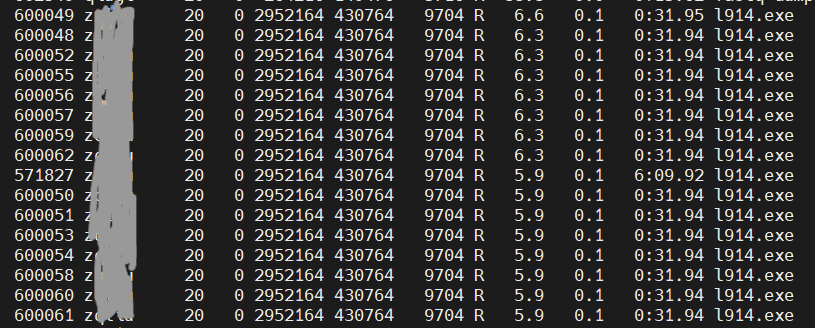
图中,任务相关进程16个(---ntasks-per-node=16),但每个任务只有6%左右的使用率,说明没有利用好cpu。
gpu利用率¶
申请资源如下:
#SBATCH --nodes=1
#SBATCH -c 9
#SBATCH --gres=gpu:1
gpu使用率如下:
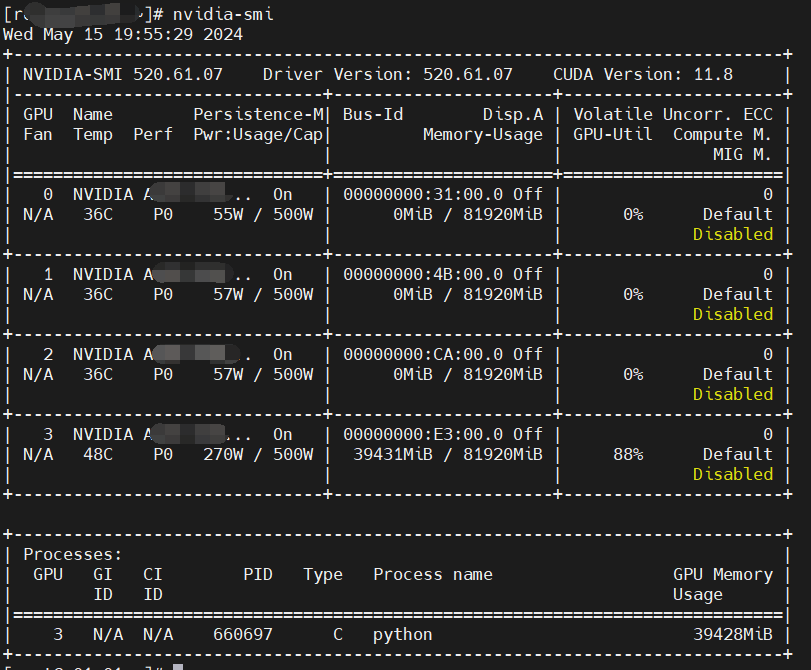
图中,脚本使用了一个gpu卡,其中显存使用39428M,gpu利用率88%。
节点数,cpu或者gpu的数量的选择¶
多线程下的并行效率¶
一些软件,如NumPy和MATLAB中的线性代数例程,能够通过使用共享内存并行编程模型(如OpenMP、英特尔线程构建块(TBB)或pthreads)编写的库使用多个CPU内核。 对于纯多线程代码,只能使用单个节点和单个任务(即nodes=1和ntasks=1),然后寻求每个任务的cpus的最佳值:
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=<T>
请参阅多线程作业的Slurm脚本示例。为了找到<T>的最佳值,我们使用了集群1中/share/case/gaussian/gaussianJob1目录下的脚本进行了如下的分析, 其中每一次任务的CPU是不同的,并且记录每个任务的运行时间:
节点数 |
cpus-per-task |
运行时间(秒) |
加速比 |
并行效率 |
|---|---|---|---|---|
1 |
1 |
15119 |
1.00 |
100% |
1 |
2 |
7552 |
2.00 |
100% |
1 |
4 |
3842 |
3.94 |
98% |
1 |
8 |
1979 |
7.64 |
95% |
1 |
16 |
963 |
15.70 |
98% |
1 |
24 |
803 |
18.83 |
78% |
1 |
32 |
764 |
19.79 |
62% |
1 |
64 |
784 |
19.28 |
30% |
在上表中,执行时间是作业运行所需的时间,加速比是串行作业的执行时间(每个任务的CPU=1)除以执行时间。 并行效率是相对于串联作业时间进行测量的。也就是说,对于每个任务的cpu=2,我们有15119.0/(7552.0×2)=100%。
上表中的数据揭示了两个关键点:
1.执行时间随着CPU内核数量的增加而减少,直到运行64个cpu的时候,代码运行速度比使用32个内核时还慢,此时可以看到cpu利用率最高也就达到2500%。这表明我们的目标不是使用尽可能多的CPU内核,而是找到最佳值。
2.从并行效率看,每个任务的cpu核数的可以是2、4、8或16,最佳值16。不考虑32个核或者64个CPU核,因为并行效率太低,。
在这种情况下,Slurm脚本可能会使用以下脚本:
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=16
此时既能保证并行效率,也使计算速度相对来说是最快的。
多节点/并行MPI作业的并行效率¶
例如,对于使用MPI的多节点代码,您需要改变每个节点的节点数和ntask。如果使用单个节点时并行效率非常高,则仅使用一个以上的节点。要最大限度地缩短完成时间,请选择能提供合理加速的最小Slurm参数集。 对于不使用线程的纯MPI代码(例如,OpenMP),每个任务的cpus=1,目标是找到每个节点的节点和ntask的最佳值:
#SBATCH --nodes=<M>
#SBATCH --ntasks-per-node=<N>
#SBATCH --cpus-per-task=1
以下是并行MPI代码的分析示例(来源于集群1中/share/case/vasp/vaspJob1目录下的作业):
节点数 |
ntasks-per-node |
CPU核数 |
运行时间(秒) |
加速比 |
并行效率 |
|---|---|---|---|---|---|
1 |
1 |
1 |
1173 |
1.00 |
100% |
1 |
2 |
2 |
631 |
1.86 |
93% |
1 |
4 |
4 |
341 |
3.44 |
86% |
1 |
8 |
8 |
182 |
6.45 |
81% |
1 |
16 |
16 |
104 |
11.28 |
70% |
1 |
32 |
32 |
77 |
15.23 |
48% |
1 |
64 |
64 |
54 |
21.72 |
34% |
2 |
64 |
128 |
42 |
27.93 |
22% |
我们看到,在使用8个ntasks之前,代码的性能还算良好。一个好的选择可能是使用8个ntasks,其中并行效率还有81%。如果对执行时间要求高一些,可选择16核,此时并行效率仍然有70%。
多节点多线程作业的并行效率¶
有些代码同时利用了共享和分布式内存并行(例如OpenMP和MPI)。在这些情况下,你需要更改--nodes、--ntasks-per-node和--cpus-per-task。 如上所述构造一个表,但每个任务包含一个新的cpus列。在使用整个节点时,--ntasks-per-node和--cpus-per-task的乘积应等于每个节点的CPU核总数。
#SBATCH --nodes=<M>
#SBATCH --ntasks-per-node=<N>
#SBATCH --cpus-per-task=<T>
通过表来查找这些Slurm参数的最佳值:
GPU作业¶
在考虑多个GPU之前,应首先证明使用单个GPU时GPU的高利用率。 如果GPU利用率对于单个GPU来说足够高,那么你应该如上述例子构造表来分析多个GPU情况下作业并行效率。
错误示例¶
示例1
#!/bin/bash
#SBATCH --job-name python_job
#SBATCH --nodes=1 # 节点数
#SBATCH --ntasks-per-node=1 # 每个节点的任务数
#SBATCH -p cpuXeon6458
#SBATCH -c 20
module load anaconda/3-2024.02.01
conda activate biotree
python demo.py
conda deactivate
此脚本设置一个进程,20个cpu,用多线程来跑python代码。首先需要确定代码是否支持多线程,另外据了解,python对于多线程的支持并不太好,可以尝试用多进程并行的办法。
示例2
#!/bin/bash
#SBATCH --job-name cp2k_job
#SBATCH --partition cpuXeon6458
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=64
module load cp2k/2024.1
mpirun -n 10 cp2k.psmp -i Ac.inp -o test.out
此脚本申请了64个cpu,但是使用mpirun的时候只设置了10个,造成资源浪费。
示例3
#!/bin/bash
#SBATCH --job-name cp2k_job
#SBATCH --partition cpuXeon6458
#SBATCH --nodes=4
#SBATCH --ntasks-per-node=16
module load cp2k/2024.1
mpirun -n 64 cp2k.psmp -i Ac.inp -o test.out
此脚本使用4个节点跑脚本,每个节点16个进程(一个进程对应一个cpu),但建议尽量将程序运行在同一节点上(设置为-N 1,-n 64),以提高计算性能。
Contributor:mzliu