GROMACS¶
简介¶
GROMACS是一种分子动力学应用程序,可以模拟具有数百至数百万个粒子的系统的牛顿运动方程。 GROMACS旨在模拟具有许多复杂键合相互作用的生化分子,例如蛋白质,脂质和核酸。
安装列表¶
序号 |
集群 |
平台 |
版本 |
编译选项 |
模块名 |
安装位置 |
|---|---|---|---|---|---|---|
1 |
hpckapok1 |
cpu |
2024.2 |
gcc11.4.0,openmpi5.0.3 |
gromacs/2024.2-gcc-openmpi5-cpu |
/share/software/gromacs/2024.2-gcc-openmpi5-cpu |
2 |
hpckapok1 |
cpu |
2023.3 |
oneapi2023.2.0 |
gromacs/2023 |
/share/software/gromacs/2023.3-oneapi |
3 |
hpckapok1 |
GPU |
2024.2 |
gcc,openmpi,cuda11.8 |
gromacs/2024.2-gcc-openmpi5-gpu |
/share/software/gromacs/2024.2-gcc-openmpi5-GPU-aware |
4 |
hpckapok2 |
cpu |
2021.5 |
oneapi2021.3 |
apps/gromacs/intelmpi/2021.5 |
/public/software/apps/gromacs/intelmpi/2021.5 |
5 |
hpckapok2 |
cpu |
2024.2 |
gcc,openmpi |
apps/gromacs/openmpi/2024.2-gcc-cpu |
/public/software/apps/gromacs/openmpi/2024.2-gcc-cpu |
6 |
hpckapok2 |
GPU |
2024.2 |
gcc,openmpi,cuda12.3 |
apps/gromacs/openmpi/2024.2-gcc-GPU-aware |
/public/software/apps/gromacs/openmpi/2024.2-gcc-gpu-aware |
使用gmx_mpi -version可以查看编译选项,比如:
$ /share/software/gromacs/2024.2-gcc-openmpi5-GPU-aware/gmx_mpi -version
:-) GROMACS - gmx_mpi, 2024.2 (-:
Executable: /share/software/gromacs/2024.2-gcc-openmpi5-GPU-aware/bin/./gmx_mpi
Data prefix: /share/software/gromacs/2024.2-gcc-openmpi5-GPU-aware
Working dir: /share/software/gromacs/2024.2-gcc-openmpi5-GPU-aware/bin
Command line:
gmx_mpi -version
GROMACS version: 2024.2
Precision: mixed
Memory model: 64 bit
MPI library: MPI
MPI library version: Open MPI v4.1.5, package: Open MPI abuild@obs-worker1 Distribution, ident: 4.1.5, repo rev: v4.1.5, Feb 23, 2023
OpenMP support: enabled (GMX_OPENMP_MAX_THREADS = 128)
GPU support: CUDA
NBNxM GPU setup: super-cluster 2x2x2 / cluster 8
... ...
使用方法¶
hpckapok1¶
cpu版(2024.2-gcc-openmpi5-cpu)¶
#!/bin/bash
#SBATCH --job-name gromacs_job
#SBATCH --partition cpuXeon6458
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=16
module load gcc/11.4.0
module swap openmpi4 mpi/openmpi/5.0.3_gnu
module load gromacs/2024.2-gcc-openmpi5-cpu
#或者用如下命令加载gromacs
#source /share/software/gromacs/2024.2-gcc-openmpi5-cpu/bin/GMXRC
gmx_mpi grompp -f md.mdp -c npt.gro -r npt.gro -t npt.cpt -p topol.top -n index.ndx -maxwarn 6 -o md_0_1.tpr
mpirun -np 16 gmx_mpi mdrun -v -deffnm md_0_1
cpu版(2023)¶
#!/bin/bash
#SBATCH --job-name gromacs_job
#SBATCH --partition cpuXeon6458
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=16
module load oneapi/2024.0
module load gromacs/2023
#或者用如下命令加载gromacs
#source /share/software/gromacs/2023.3-oneapi/bin/GMXRC
gmx_mpi grompp -f md.mdp -c npt.gro -r npt.gro -t npt.cpt -p topol.top -n index.ndx -maxwarn 6 -o md_0_1.tpr
mpirun -np 16 gmx_mpi mdrun -v -deffnm md_0_1
gpu版(2024.2-gcc-openmpi5-gpu)¶
#!/bin/bash
#SBATCH --job-name gromacs_job
#SBATCH --partition gpuA800
#SBATCH --nodes=1
#SBATCH --gres=gpu:1
#SBATCH --ntasks-per-node=9
module load cuda/12.3
module swap openmpi4 mpi/openmpi/5.0.3
module load gromacs/2024.2-gcc-openmpi5-gpu
#或者用如下命令加载gromacs
#source /share/software/gromacs/2024.2-gcc-openmpi5-GPU-aware/bin/GMXRC
gmx_mpi grompp -f md.mdp -c npt.gro -r npt.gro -t npt.cpt -p topol.top -n index.ndx -maxwarn 6 -o md_0_1.tpr
mpirun -np 9 gmx_mpi mdrun -nb gpu -v -deffnm md_0_1
集群1的算例可以在/share/case/gromacs/gromacsJob1中找到。
hpckapok2¶
cpu版本(2024.2-gcc-cpu)¶
#!/bin/bash
#SBATCH -J gromacs_job
#SBATCH -p cpuXeon6458
#SBATCH -N 1
#SBATCH -n 64
module load compiler/gcc/11.4.0
module load mpi/openmpi/gnu/5.0.3
module load apps/gromacs/openmpi/2024.2-gcc-cpu
#或者用如下命令加载gromacs
#source /public/software/apps/gromacs/openmpi/2024.2-gcc-cpu/bin/GMXRC
mpirun -np 64 gmx_mpi mdrun -v -deffnm nvt
cpu版本(intelmpi/2021.5)¶
#!/bin/bash
#SBATCH -J gromacs_job
#SBATCH -p cpuXeon6458
#SBATCH -N 1
#SBATCH -n 64
module load compiler/intel/2021.3.0
module load mpi/intelmpi/2021.3.0
module load apps/gromacs/intelmpi/2021.5
#或者用如下命令加载gromacs
#source /public/software/apps/gromacs/intelmpi/2021.5/bin/GMXRC
mpirun -np 64 gmx_mpi mdrun -v -deffnm nvt
gpu版本(2024.2-gcc-GPU-aware)¶
#!/bin/bash
#SBATCH -J gromacs_job
#SBATCH -p gpuA800
#SBATCH -N 1
#SBATCH --gres=gpu:1
#SBATCH -n 8
module load compiler/gcc/11.4.0
module load mpi/openmpi/gnu/5.0.3
module load cuda/12.3.0
module load apps/gromacs/openmpi/2024.2-gcc-GPU-aware
#或者用如下命令加载gromacs
#source /public/software/apps/gromacs/openmpi/2024.2-gcc-gpu-aware/bin/GMXRC
mpirun -np 8 gmx_mpi mdrun -v -deffnm nvt -nb gpu
集群2的算例可以在/public/software/share/case/gromacs/gmx-1找到。
SCOW平台¶
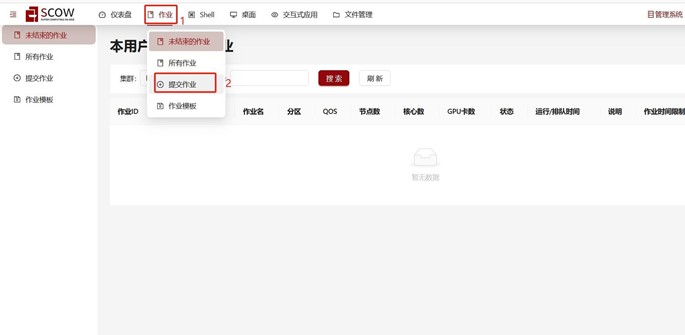
Step 1. 在scow平台打开作业,点击提交作业
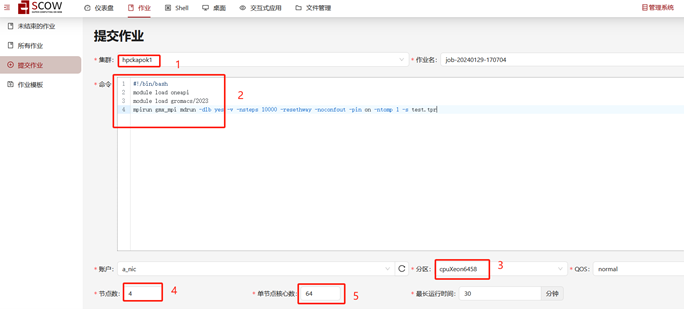
Step 2. 在提交作业界面,选择集群分区,填写相应代码作业和指向测试文件,选择节点和核心
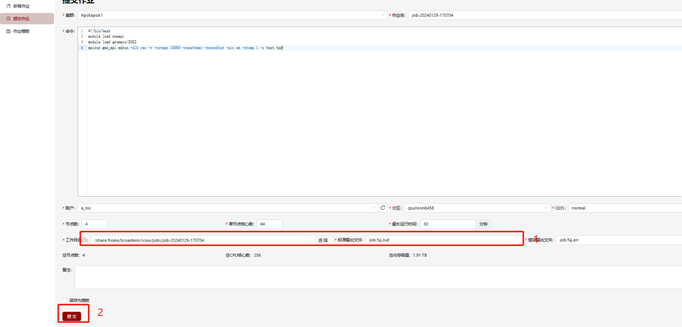
Step 3. 填写输出文件位置,点击提交作业
参考资料¶
Contributor:B君、mzliu